Amplicon data analysis - workflow¶
This page descibes the usual steps in an amplicon data analysis after you have obtained an ASV-table.
Quality control¶
First step of an analysis is always to ensure that the data is of sufficient quality. Below are descibed the four main focus points of such a quality check.
Check composition¶
Usually we have prior knowledge on which microbes we find in a given environment. Check if you find the expected microbes in your sample, or if your fecal samples are filled with common soil-dwelling bacteria. This should be done a per-sample basis - maybe a few samples are completely off and should be discarded. You would also create rarefaction curves (see notebook on Compositionality) to check if any samples have too low read depth to cover the diversity.

Positive controls¶
For each sequencing run we always include a mock community, which is DNA from different microbes mixed with a known composition. The composition of this sample should of course match the expected to a reasonable degree. Another positive control which could be added, but rarely is, would be to grow a selection of microbes in the lab and mix them to a known composition. The latter differs from the former type of control in that it also includes DNA extraction bias.
As an example the figure below from the paper 16S rRNA gene sequencing of mock microbial populations- impact of DNA extraction method, primer choice and sequencing platform shows how different the sequenced composition is from the expected composition. DNA mock community samples usually match much better with the expected, but are also ignoring DNA extraction bias, which is known to be substantial.
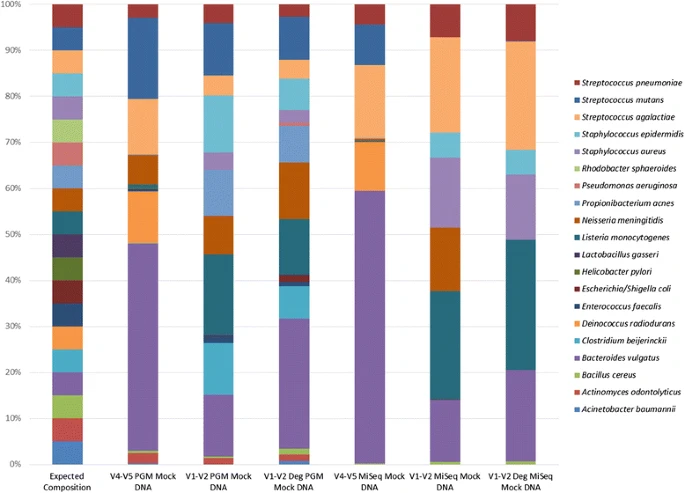
Negative controls¶
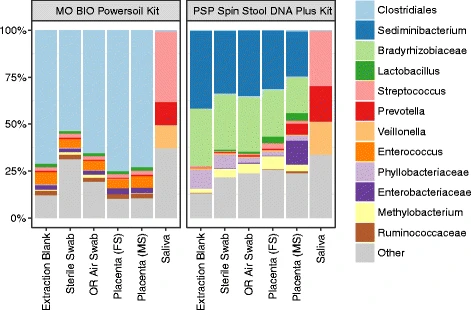
We always include three different negative controls: Water added at the DNA extraction step, water added at the 1st PCR, and water added at the 2nd PCR. A fourth that could be added, depending on the study design, is a sampling negative control; for example if samples were collected with "sterile" cotton swaps, then you would also include an unused swap for DNA extraction and sequencing to ensure that there was no foreign DNA present in the swaps themselves. In an ideal world these were all completely devoid of microbes, but lab reagants contain tiny amounts of bacteria or their DNA (also called the kitome) which will get amplified during PCR. The sample can also be contaminated from the lab environment or from nearby samples when handled on a 96-well plate (cross-contamination or splashome). The composition of the negative controls should differ considerably from the actual samples. Often contamination is worst when samples have low biomass - in this case utmost care should be taken during handling in the lab. If there is compositional overlap between actual samples and negative controls one mitigation could be decontaminaiton during data analysis, where taxa that are found in negative controls are removed from the dataset across all samples. However, if your actual samples contain microbes that are commonly found in lab reagants, other measures should be taken to ensure that the data is trustworthy.
The importance of negative controls is vizualized in this figure from Optimizing methods and dodging pitfalls in microbiome research where we can see that the Placenta samples are hardly distinguishable from the 3 negative controls:
Technical variation - batch effects¶
Lastly, one should check whether the composition correlates with any technical covariants (which it shouldn't). Technical covariates could be if DNA extraction was done on different days or by different people or with different batches of kit. Similarly, if samples are from different sequencing runs, were stored for different durations, or otherwise handled differently during the process from sampling to sequencing, this should be ensured not the correlate with the microbial composition. Because these technical differences can create variation in the resulting data it is crucial to randomize samples during lab handling if possible. It would be a big problem you were running a treatment/control experiment and extracted DNA from all the control samples on one day, and all the treatment samples on another day. In this case you would not be able to distiguish between variation between treatment/control groups and day-to-day variation in DNA extraction efficiency/bias. If there is technical variation one should control for it during data analysis or ensure that it doesn't affect the results.
Compositional transformation, rarefaction, or other normalization?¶
After quality control, one would usually decide the normalization and read-depth-bias strategy. Either rarefy the data and use the rarefied dataset throughout or rely on other ways of controlling for read depth bias. See details in notebook on compositionality. The choice matters. See figure 4 from Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible comparing analytical accuracy with different normalizations:
Analysis¶
There is no one-size-fits-all analysis. The choices depend entirely on the aim and hypotheses of your study:
- Will you test for differences in diversity between samples, use alpha diversity
- Will you test for differences in overall composition between samples, use beta diversity
- Do you hypothesize that specific microbes have different abundances across your samples, use differential abundance
- Do you want to vizualize which microbes are present in your samples, use pretty heatmaps, relative abundance bar charts, or phylogenetic trees.
- Do you want to identify a core microbiome or microbes specific to groups of samples, make a venn diagram
- Are you interested in predicting the origin of samples depending on the microbes present, use supervised machine learning
- Do you think your data is separated into different groups, use clustering
- Are you interested in how microbes co-vary with each other, use microbial association networks
and the list could go on...